FastCCC’s Inputs for Reference-based Analyses
Introduction
The functions infer_query_workflow requires three input datasets:
- ScRNA-seq data (raw counts are stored in compressed sparse row (
CSR) format within an.h5adfile.). - Cell type annotations (provided as a tab-separated table or as a column in obsm of the .h5ad file).
- Ligand-receptor interaction (LRI) database (precompiled datasets are available for direct use).
- Constructed reference panel (precompiled reference are available for direct use).
For additional parameters, refer to the Function section. For specific usage examples, refer to the Basic Usage section and Code Snippets.
1. ScRNA-seq data
An .h5ad file readable by Scanpy, containing raw scRNA-seq counts stored in compressed sparse row (CSR) format. To compare with reference panel, FastCCC requires this format and will preprocess the query dataset using the same workflow as for the reference data.
Please avoid gene filtering, as FastCCC quantifies and ranks the expression levels of all genes in each cell. In this context, no additional preprocessing of scRNA-seq data is necessary.
Ensure that HGNC symbols (official gene symbols) are used as gene names to maintain consistency with the reference panel. (In future versions, we plan to introduce a gene ID conversion feature for easier usage—stay tuned.)
The internal matrix structure of the scRNA-seq data appears as follows:
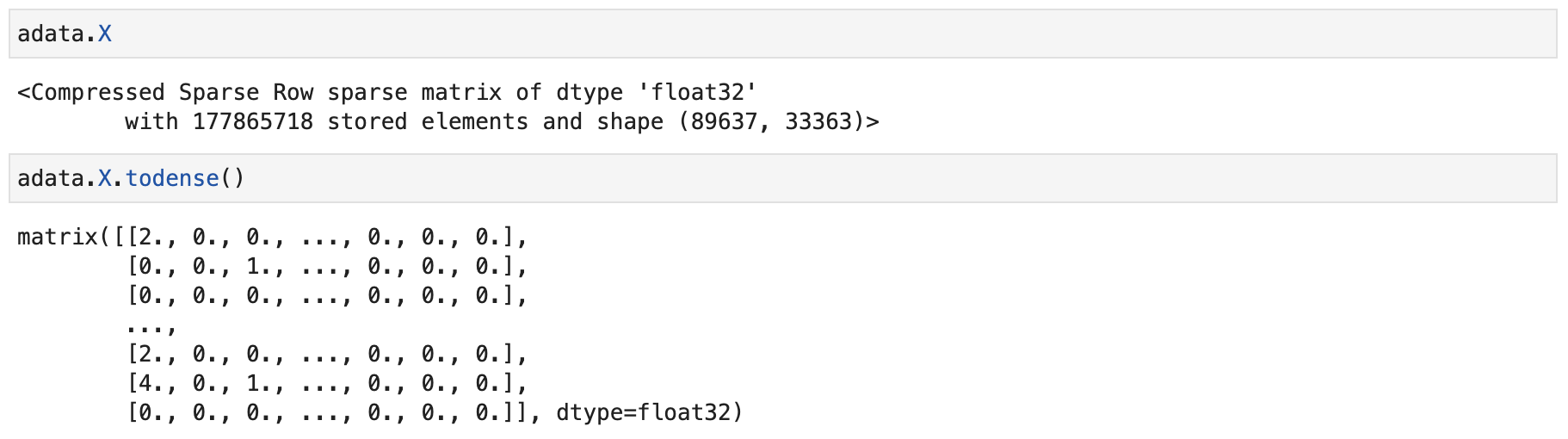
Here is an example of how to construct a suitable input if your data does not meet the required format.
2. Cell type annotations
For ease of use, cell type annotations can be provided in two formats.
- A TSV (tab-separated) file containing two columns—one for cell IDs (matching those in the .h5ad file) and another for cell type labels. (see example)
- Directly using the cell type annotations stored in the .h5ad file’s obs field (if available). In this case, specify the corresponding column name using the meta_key parameter. (see example)
FastCCC can analyze cell types that are not present in the reference panel. However, to better leverage the accumulated information in the reference dataset, if a cell type in the query dataset corresponds to an existing cell type in the reference but has a different string label, you can use celltype_mapping_dict to map them accordingly.
celltype_mapping_dict = {
# cell type in reference : cell type in query
"endothelial cell of lymphatic vessel": "Endothelial Cells",
"naive B cell": "B Cells",
"class switched memory B cell": "B Cells",
"IgG plasma cell": "Plasma Cells",
"IgA plasma cell": "Plasma Cells"
}
The cell type information contained in the reference panel can be found in the file:
your_path/reference/tissue/config.toml
This file provides an overview of the cell types included in the reference panel, helping users align their query dataset accordingly.

Refer to the Functions and Example2 sections for more details.
3. Ligand-receptor interaction database
We provide preprocessed database on GitHub for users to download directly.
Refer to the Basic usage - Inputs section for more details.
4. Constructed reference panel
We provide preprocessed database on GitHub for users to download directly. A complete reference database includes the following components:
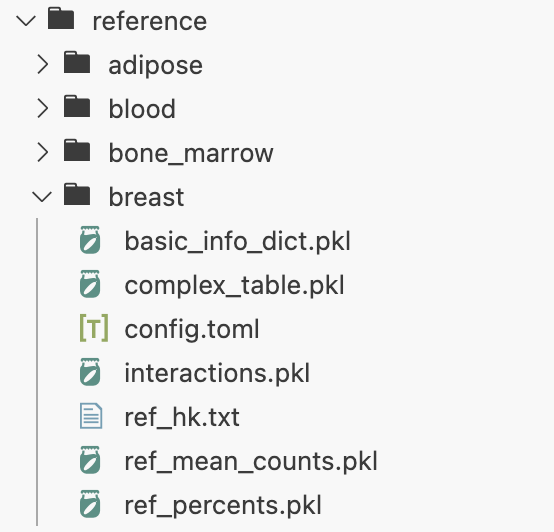
To build your own reference panel, see here.
For additional parameters, refer to the Function section.